AAV-cTNT尾靜脈注射小鼠心臟的病毒用量的研究_abio生物試劑品牌網
心力衰竭(HF)是全球主要死因之一,目前治療手段有限。環磷酸腺苷(cAMP)在心臟的正常和病理信號傳導中起著重要作用。PDE4是心臟中降解cAMP的主要磷酸二酯酶,PDE4的4B和4D亞型在心臟功能調節中的作用存在爭議。PDE4B過表達對心臟重塑和HF有益,然而,PDE4D和PDE4抑制劑在心力衰竭中的作用仍不明確。2025年2月22日,華中科技大學同濟醫學院基礎醫學院付琴教授團隊在Redox Biology?(IF10.7) 發文“PDE4D inhibition ameliorates cardiac hypertrophy and heart fAIlure by activating mitophagy”。該研究發現 PDE4D 在心力衰竭中表達增加,抑制PDE4D可通過激活線粒體自噬改善心肌肥厚和心力衰竭,為心力衰竭治療提供新靶點和策略。
 · 維真助力·
· 維真助力·
基因信息:PDE4D5:磷酸二酯酶4D變體
實驗動物:PDE4D+/?及WT小鼠
病毒產品:AAV9-cTNT-PDE4D5、AAV9-NC
注射方式:尾靜脈注射
病毒用量:5.0x10^11vg/mouse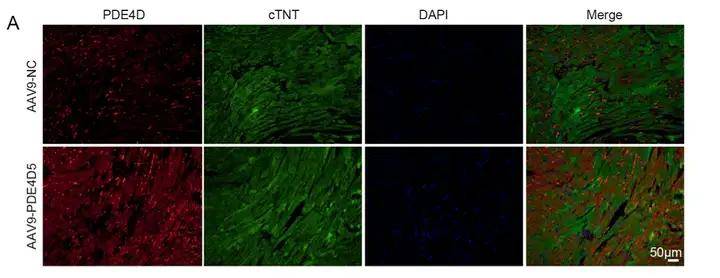 免疫熒光染色檢測PDE4D在心肌細胞中特異性過表達
研究結果
免疫熒光染色檢測PDE4D在心肌細胞中特異性過表達
研究結果
1、PDE4D誘導心肌細胞肥大和氧化應激
體內實驗數據顯示使用PDE4抑制劑羅氟司特可抑制異丙腎上腺素誘導的心肌肥大和心力衰竭,同時ISO處理使小鼠心臟PDE4D mRNA和蛋白水平上調,并降低PKA 活性;羅氟司特處理可抑制PDE4D的誘導,使PDE4活性、cAMP含量以及CREB和受磷蛋白的磷酸化水平恢復正常,且對PDE4A和PDE4B的表達無顯著影響。體外實驗進一步驗證了PDE4抑制劑的心臟保護作用,通過對比PDE4D 和PDE4B對心肌細胞的不同影響,發現PDE4D可誘導心肌細胞肥大和氧化應激,而PDE4B無此作用。與非衰竭心臟相比,衰竭人類心臟中PDE4D蛋白表達升高,其中 PDE4D5 變體表達顯著增加。通過在NRVMs中過表達PDE4D5,發現PDE4D5可能參與心臟肥大和線粒體損傷的病理過程。進一步研究證實PDE4D5過表達會抑制心肌細胞中PINK1和Parkin的蛋白表達,降低LC3BII/I比值,增加P62表達,而PDE4B過表達對上述指標無明顯影響,綜合說明線粒體自噬受損與PDE4D誘導的心肌細胞肥大和線粒體損傷有關。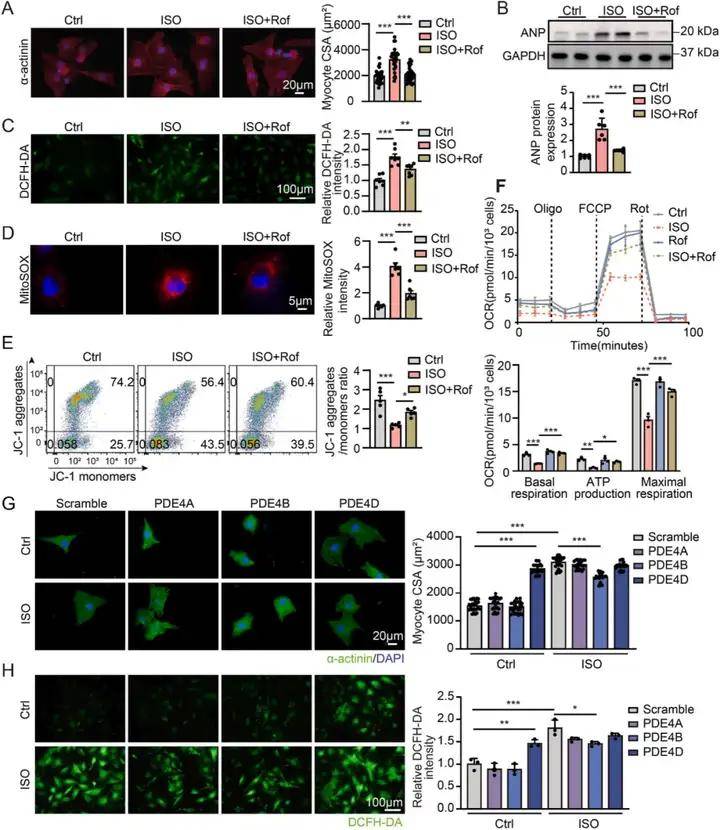 PDE4D,而不是PDE4B,誘導心肌細胞肥大和氧化應激
2、PDE4抑制劑通過SIRT1/PINK1/Parkin通路誘導線粒體自噬,保護心肌細胞免受肥大和氧化應激
PDE4D,而不是PDE4B,誘導心肌細胞肥大和氧化應激
2、PDE4抑制劑通過SIRT1/PINK1/Parkin通路誘導線粒體自噬,保護心肌細胞免受肥大和氧化應激
使用siRNA敲低PINK1表達后,羅氟司特誘導的PINK1和Parkin表達增加被消除,其對ISO處理的NRVMs 細胞大小、氧化應激和線粒體損傷的保護作用也被阻斷,且抑制了羅氟司特誘導的Parkin和LC3B與線粒體的共定位。SIRT1可通過改善PINK1/Parkin介導的線粒體自噬保護線粒體。ISO處理顯著降低心肌細胞中SIRT1的表達,羅氟司特可減弱這一降低。用SIRT1 shRNA處理后,羅氟司特對ISO誘導的心臟肥大、氧化應激和線粒體損傷的保護作用被消除,其誘導的PINK1、Parkin和LC3B與線粒體的共定位也減弱,表明PDE4抑制劑通過激活SIRT1介導的PINK1/Parkin 信號通路誘導線粒體自噬,發揮心臟保護作用。進一步研究表明PDE4 抑制劑可減輕TAC誘導的心臟肥大和心力衰竭,主要通過促進心臟線粒體自噬實現。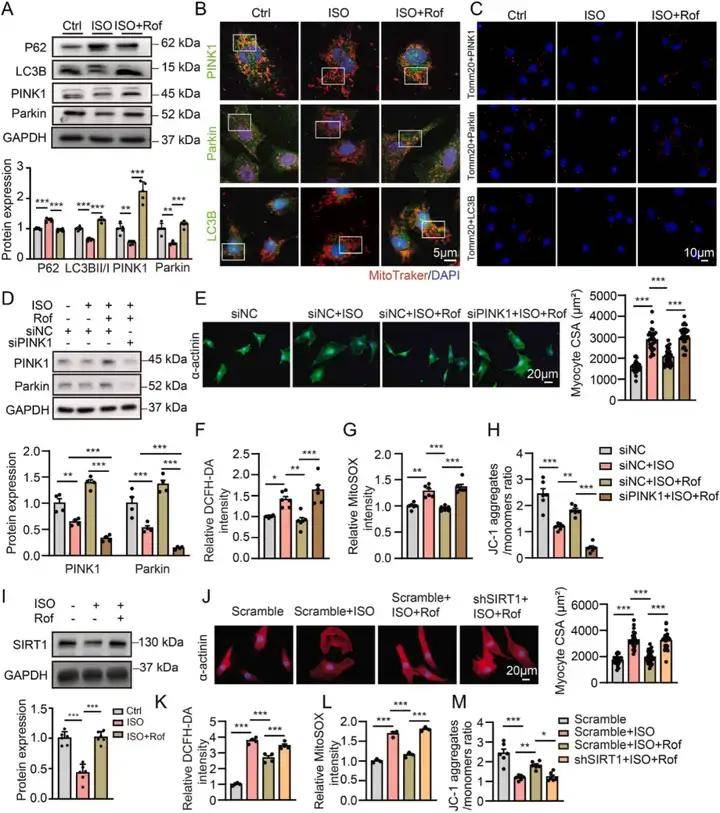 PDE4抑制劑通過SIRT1激活誘導線粒體自噬,防止心肌細胞肥大和線粒體功能障礙
3、PDE4D5的心臟過表達抵消了PDE4D敲除對TAC小鼠的心臟保護作用
PDE4抑制劑通過SIRT1激活誘導線粒體自噬,防止心肌細胞肥大和線粒體功能障礙
3、PDE4D5的心臟過表達抵消了PDE4D敲除對TAC小鼠的心臟保護作用
選用他莫昔芬誘導的心臟特異性雜合PDE4D敲除(PDE4DhCKO )小鼠,進行TAC手術及他莫昔芬處理,通過評估心臟功能、細胞狀態及線粒體自噬情況,證實下調PDE4D對TAC誘導的心臟肥大和心力衰竭有保護作用。為了進一步研究PDE4D5在體內病理性心肌肥大和心力衰竭中的作用,研究團隊使用雜合PDE4D基因敲除小鼠(PDE4D+/-),通過靜脈注射攜帶PDE4D5的AAV9載體,使心臟特異性過表達PDE4D5,這一操作加劇了TAC小鼠的心臟肥大和收縮功能障礙,同時惡化了細胞凋亡、ROS水平和MDA含量,還降低了線粒體嵴評分、線粒體自噬體形成和ATP含量。此外,PDE4D5 過表達抵消了PDE4D+/?小鼠因PDE4D基因敲除所產生的保護效果,表明PDE4D5在促進心臟肥大和心力衰竭中起到關鍵作用,它主要通過抑制線粒體自噬來實現這一過程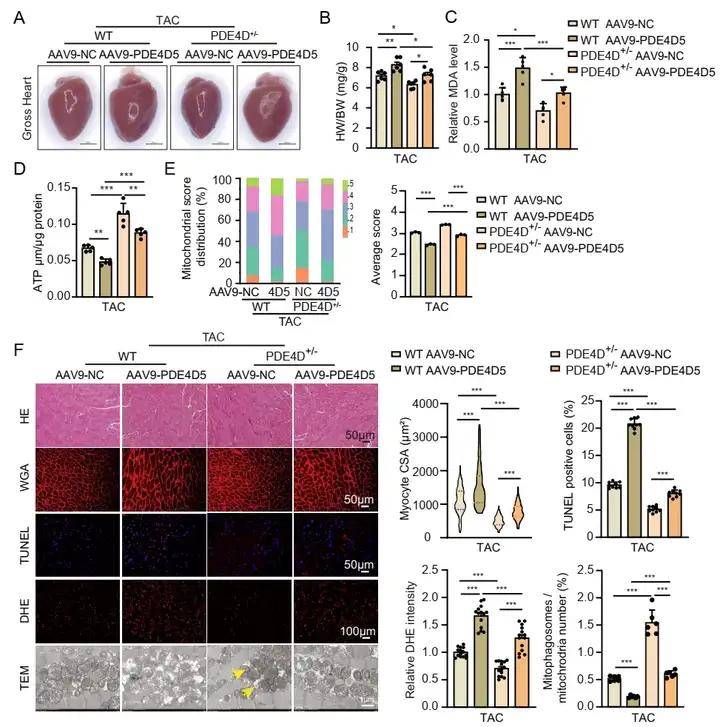 PDE4D5的心臟過表達抵消了PDE4D敲除對TAC小鼠的心臟保護作用
研究結論
PDE4D5的心臟過表達抵消了PDE4D敲除對TAC小鼠的心臟保護作用
研究結論
本研究闡明持續腎上腺素能激活通過cAMP-PKA信號上調PDE4D表達,抑制 CREB-SIRT1信號介導的線粒體自噬,導致心肌細胞肥大和線粒體損傷。強調抑制PDE4D表達可能是治療HF的一種新策略。
· 維真助力·基因信息:PDE4D5:磷酸二酯酶4D變體
實驗動物:PDE4D+/?及WT小鼠
病毒產品:AAV9-cTNT-PDE4D5、AAV9-NC
注射方式:尾靜脈注射
病毒用量:5.0x10^11vg/mouse
免疫熒光染色檢測PDE4D在心肌細胞中特異性過表達
研究結果1、PDE4D誘導心肌細胞肥大和氧化應激
體內實驗數據顯示使用PDE4抑制劑羅氟司特可抑制異丙腎上腺素誘導的心肌肥大和心力衰竭,同時ISO處理使小鼠心臟PDE4D mRNA和蛋白水平上調,并降低PKA 活性;羅氟司特處理可抑制PDE4D的誘導,使PDE4活性、cAMP含量以及CREB和受磷蛋白的磷酸化水平恢復正常,且對PDE4A和PDE4B的表達無顯著影響。體外實驗進一步驗證了PDE4抑制劑的心臟保護作用,通過對比PDE4D 和PDE4B對心肌細胞的不同影響,發現PDE4D可誘導心肌細胞肥大和氧化應激,而PDE4B無此作用。與非衰竭心臟相比,衰竭人類心臟中PDE4D蛋白表達升高,其中 PDE4D5 變體表達顯著增加。通過在NRVMs中過表達PDE4D5,發現PDE4D5可能參與心臟肥大和線粒體損傷的病理過程。進一步研究證實PDE4D5過表達會抑制心肌細胞中PINK1和Parkin的蛋白表達,降低LC3BII/I比值,增加P62表達,而PDE4B過表達對上述指標無明顯影響,綜合說明線粒體自噬受損與PDE4D誘導的心肌細胞肥大和線粒體損傷有關。
PDE4D,而不是PDE4B,誘導心肌細胞肥大和氧化應激
2、PDE4抑制劑通過SIRT1/PINK1/Parkin通路誘導線粒體自噬,保護心肌細胞免受肥大和氧化應激使用siRNA敲低PINK1表達后,羅氟司特誘導的PINK1和Parkin表達增加被消除,其對ISO處理的NRVMs 細胞大小、氧化應激和線粒體損傷的保護作用也被阻斷,且抑制了羅氟司特誘導的Parkin和LC3B與線粒體的共定位。SIRT1可通過改善PINK1/Parkin介導的線粒體自噬保護線粒體。ISO處理顯著降低心肌細胞中SIRT1的表達,羅氟司特可減弱這一降低。用SIRT1 shRNA處理后,羅氟司特對ISO誘導的心臟肥大、氧化應激和線粒體損傷的保護作用被消除,其誘導的PINK1、Parkin和LC3B與線粒體的共定位也減弱,表明PDE4抑制劑通過激活SIRT1介導的PINK1/Parkin 信號通路誘導線粒體自噬,發揮心臟保護作用。進一步研究表明PDE4 抑制劑可減輕TAC誘導的心臟肥大和心力衰竭,主要通過促進心臟線粒體自噬實現。
PDE4抑制劑通過SIRT1激活誘導線粒體自噬,防止心肌細胞肥大和線粒體功能障礙
3、PDE4D5的心臟過表達抵消了PDE4D敲除對TAC小鼠的心臟保護作用選用他莫昔芬誘導的心臟特異性雜合PDE4D敲除(PDE4DhCKO )小鼠,進行TAC手術及他莫昔芬處理,通過評估心臟功能、細胞狀態及線粒體自噬情況,證實下調PDE4D對TAC誘導的心臟肥大和心力衰竭有保護作用。為了進一步研究PDE4D5在體內病理性心肌肥大和心力衰竭中的作用,研究團隊使用雜合PDE4D基因敲除小鼠(PDE4D+/-),通過靜脈注射攜帶PDE4D5的AAV9載體,使心臟特異性過表達PDE4D5,這一操作加劇了TAC小鼠的心臟肥大和收縮功能障礙,同時惡化了細胞凋亡、ROS水平和MDA含量,還降低了線粒體嵴評分、線粒體自噬體形成和ATP含量。此外,PDE4D5 過表達抵消了PDE4D+/?小鼠因PDE4D基因敲除所產生的保護效果,表明PDE4D5在促進心臟肥大和心力衰竭中起到關鍵作用,它主要通過抑制線粒體自噬來實現這一過程
PDE4D5的心臟過表達抵消了PDE4D敲除對TAC小鼠的心臟保護作用
研究結論本研究闡明持續腎上腺素能激活通過cAMP-PKA信號上調PDE4D表達,抑制 CREB-SIRT1信號介導的線粒體自噬,導致心肌細胞肥大和線粒體損傷。強調抑制PDE4D表達可能是治療HF的一種新策略。
本站“ABIO生物試劑品牌網”圖片文字來自互聯網
如果有侵權請聯系微信: nanhu9181 處理,感謝~